Timon Harz
December 16, 2024
Microsoft and Novartis Introduce Chimera: AI Framework for Scalable and Accurate Retrosynthesis Prediction
Chimera combines diverse machine learning models to predict synthetic routes with unprecedented accuracy, revolutionizing chemical synthesis planning. This collaboration is set to reshape the future of AI-driven chemistry, offering a scalable solution for complex molecular design.

Chemical synthesis plays a pivotal role in creating new molecules for medicine, materials science, and fine chemicals. This complex process involves designing chemical reactions to produce specific target molecules and has historically depended on human expertise. However, recent advancements in computational methods have revolutionized retrosynthesis—the strategy of working backward from a target molecule to identify the sequence of reactions needed for its synthesis. By integrating modern computational tools, researchers are addressing persistent challenges in synthetic chemistry, aiming to make retrosynthesis faster, more efficient, and highly accurate.
A major obstacle in retrosynthesis lies in predicting rare or less common chemical reactions, which are crucial for crafting innovative chemical pathways. These infrequent reactions often fall outside the scope of traditional machine-learning models due to their underrepresentation in training datasets. Additionally, multi-step retrosynthesis planning can compound errors, resulting in invalid or unfeasible synthetic routes. These limitations restrict the ability to discover diverse and creative approaches to chemical synthesis, particularly when tackling complex or novel molecular designs.
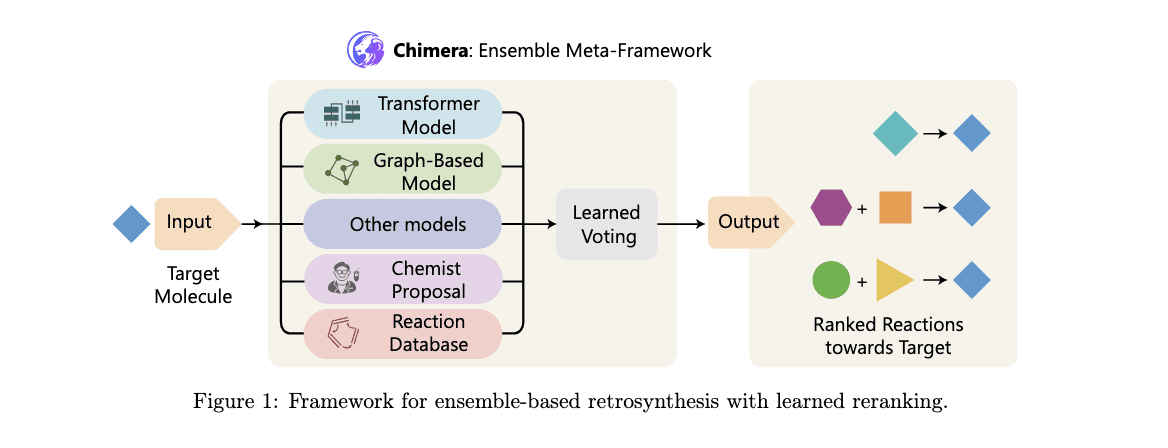
Traditional computational approaches to retrosynthesis have largely relied on single-step models or rule-based systems. These methods depend on predefined rules or extensive training datasets, which limits their ability to adapt to novel and unique reaction types. For example, graph-based and sequence-based models are commonly used to predict likely transformations. While these methods have improved the accuracy of predicting frequent reactions, they often lack the flexibility to handle the intricacies of rare and complex chemical transformations, leaving a significant gap in holistic retrosynthetic planning.
To address these challenges, researchers from Microsoft Research, Novartis Biomedical Research, and Jagiellonian University have introduced Chimera, an innovative ensemble framework for retrosynthesis prediction. Chimera combines outputs from multiple machine-learning models, each with distinct inductive biases, through a learned ranking mechanism that synthesizes their strengths. Central to this framework are two cutting-edge models: NeuralLoc, a graph neural network-based model specializing in molecule editing, and R-SMILES 2, a sequence-to-sequence Transformer model designed for de-novo retrosynthetic predictions. By integrating these advanced models, Chimera delivers enhanced accuracy and scalability, bridging critical gaps in retrosynthesis prediction.
The Chimera framework combines the outputs of its constituent models through a ranking system that assigns scores based on model agreement and predictive confidence. NeuralLoc, which encodes molecular structures as graphs, accurately predicts reaction sites and templates. This approach ensures that the predicted transformations align with established chemical rules while optimizing computational efficiency. R-SMILES 2, on the other hand, uses advanced attention mechanisms like Group-Query Attention to predict reaction pathways. Its architecture incorporates enhancements in normalization and activation functions, improving gradient flow and inference speed. By integrating these models, Chimera ranks potential reaction pathways using overlap-based scoring, effectively combining the strengths of both editing-based and de-novo approaches. This integration ensures robust predictions, even for complex or rare reactions.
Chimera’s performance has been extensively validated on several publicly available datasets, such as USPTO-50K, USPTO-FULL, and the proprietary Pistachio dataset. On the USPTO-50K dataset, Chimera outperformed previous state-of-the-art methods, achieving a 1.7% improvement in top-10 prediction accuracy. This demonstrates its ability to predict both common and rare reactions. The model further improved top-10 accuracy by 1.6% on the larger USPTO-FULL dataset. When tested on the Pistachio dataset, which is over three times larger than USPTO-FULL, Chimera maintained high accuracy across a wider array of reactions. Comparisons with organic chemists confirmed Chimera’s superiority, with its predictions consistently preferred over those from individual models, underscoring its effectiveness in real-world applications.
The framework was also evaluated on an internal Novartis dataset containing over 10,000 reactions to test its robustness under distribution shifts. In a zero-shot setting, where no additional fine-tuning was applied, Chimera outperformed its individual constituent models, demonstrating superior accuracy. This performance highlights Chimera’s ability to generalize across different datasets and predict viable synthetic pathways in real-world applications. Additionally, Chimera excelled in multi-step retrosynthesis tasks, achieving near-perfect success rates on benchmarks such as SimpRetro, far surpassing the performance of individual models. Its ability to identify pathways for highly challenging molecules further emphasizes its potential to revolutionize computational retrosynthesis.
Chimera represents a significant leap forward in retrosynthesis prediction by overcoming challenges associated with rare reaction predictions and multi-step planning. The framework’s integration of diverse models and its robust ranking mechanism enable superior accuracy and scalability. With its impressive ability to generalize across datasets and its success in tackling complex retrosynthetic tasks, Chimera is poised to drive advancements in chemical synthesis, unlocking new possibilities for molecular design and innovation.
Retrosynthesis prediction is a fundamental technique in organic chemistry that involves deconstructing complex target molecules into simpler precursor structures. This process enables chemists to design synthetic routes by identifying available starting materials and the necessary chemical reactions to assemble the desired compound. The significance of retrosynthesis prediction lies in its ability to streamline the synthesis of complex molecules, thereby accelerating the development of new pharmaceuticals, materials, and chemicals.
The Process of Retrosynthesis
The retrosynthetic approach begins with the target molecule and systematically breaks it down into simpler fragments through a series of disconnections. Each disconnection represents a potential chemical reaction that could have formed the bond in the target molecule. This recursive process continues until the fragments are simple enough to be readily available or easily synthesized. The resulting network of possible synthetic routes is known as a retrosynthetic tree, which guides chemists in selecting the most efficient and feasible pathway for synthesis.
Challenges in Retrosynthesis Prediction
Despite its utility, retrosynthesis prediction presents several challenges:
Complexity of Target Molecules: As molecules increase in complexity, the number of possible disconnections grows exponentially, making it difficult to identify the most efficient synthetic route.
Availability of Starting Materials: The ideal synthetic route should utilize commercially available or easily synthesizable starting materials. Identifying such materials requires comprehensive knowledge of chemical databases and reaction pathways.
Reaction Predictability: Not all theoretical disconnections correspond to practical or known chemical reactions. Predicting which disconnections will lead to viable reactions is a significant challenge.
Stereochemical Considerations: Maintaining the correct stereochemistry throughout the synthetic pathway is crucial, especially for biologically active compounds.
Advancements in Computational Retrosynthesis
The advent of computational chemistry and artificial intelligence has significantly advanced retrosynthesis prediction:
Reaction Databases: Extensive databases of known chemical reactions, such as the USPTO-50K dataset, have been compiled to aid in predicting feasible synthetic routes.
Machine Learning Models: Deep learning models, including graph neural networks and sequence-to-sequence architectures, have been developed to predict retrosynthetic routes by learning from large datasets of chemical reactions. These models can identify patterns and suggest plausible reaction pathways.
Template-Free Approaches: Traditional methods often rely on reaction templates, which can be limiting. Template-free approaches, such as the G2Gs framework, have been introduced to predict retrosynthetic routes without predefined templates, offering greater flexibility and scalability.
Interpretable Models: There is a growing emphasis on developing models that not only predict synthetic routes but also provide insights into the reasoning behind their predictions. This interpretability is crucial for chemists to trust and validate the suggested pathways.
Applications and Impact
The integration of AI and machine learning into retrosynthesis prediction has had a profound impact on various fields:
Pharmaceutical Development: Accelerating the design and synthesis of new drugs by predicting efficient synthetic routes, thereby reducing time and cost in drug development.
Material Science: Facilitating the creation of novel materials with desired properties by predicting synthetic pathways for complex compounds.
Sustainable Chemistry: Identifying environmentally friendly and cost-effective synthetic routes by considering factors such as reagent availability and reaction conditions.
Challenges in Traditional Retrosynthesis Prediction Methods
Retrosynthesis prediction—the process of deconstructing complex target molecules into simpler precursors—is a cornerstone of organic chemistry. Traditional methods, while foundational, face several challenges that can impede the efficiency and accuracy of synthetic route design.
1. Complexity of Target Molecules
The intricate structures of many molecules present a significant challenge. As molecular complexity increases, the number of potential disconnections grows exponentially, making it difficult to identify the most efficient synthetic route. Chemists may spend extensive time mapping out retrosynthetic routes for a single compound, which can be both time-consuming and labor-intensive.
2. Knowledge Dependency
Traditional retrosynthesis heavily relies on the chemist's expertise and knowledge of known reactions and synthetic strategies. This dependency can lead to biases and may limit the exploration of novel synthetic pathways, especially when dealing with unfamiliar or complex molecules.
3. Template-Based Limitations
Many traditional methods utilize reaction templates to predict synthetic routes. While effective for known reactions, this approach has limitations:
Limited Scope: Template-based methods are constrained by the range of reactions included in the templates, potentially overlooking novel or less common reactions.
Inflexibility: These methods may struggle to adapt to new or unforeseen reaction conditions, leading to less accurate predictions for reactions outside the template database.
4. Computational Intensity
Traditional computational methods for reaction prediction can be computationally expensive and time-consuming. They may still fail to capture the full dynamics of certain reactions, especially those involving complex mechanisms or requiring high computational resources.
5. Data Scarcity and Quality
Accurate retrosynthesis prediction relies on extensive and high-quality reaction data. Traditional methods often suffer from data scarcity, especially for rare or novel reactions. The lack of comprehensive datasets can hinder the development of robust predictive models.
6. Stereochemical Considerations
Maintaining the correct stereochemistry throughout the synthetic pathway is crucial, especially for biologically active compounds. Traditional methods may not adequately account for stereochemical factors, leading to incorrect predictions.
7. Lack of Generalization
Traditional methods may not generalize well to new or unseen reactions, limiting their applicability in diverse chemical contexts. This lack of generalization can result in inaccurate predictions when applied to novel compounds or reaction conditions.
8. Integration Challenges
Integrating various data sources and methodologies in traditional retrosynthesis prediction can be challenging. The lack of standardized protocols and data formats can hinder the development of comprehensive predictive models.
9. Limited Predictive Power
Traditional methods may struggle to predict the outcomes of complex reactions, especially those involving multiple steps or unusual reaction conditions. This limitation can lead to inefficiencies in synthetic planning and increased experimental validation.
10. Evolving Reaction Mechanisms
The dynamic nature of chemical reactions, with evolving mechanisms and intermediates, poses a challenge for traditional methods. These methods may not effectively capture the transient states and complex pathways involved in certain reactions.
In the realm of chemical synthesis, accurately predicting retrosynthetic pathways is paramount for the efficient development of new molecules, particularly in pharmaceuticals and materials science. Traditional methods, while foundational, often grapple with complexities arising from the vast diversity of chemical reactions and the intricate nature of target molecules. To address these challenges, Microsoft and Novartis have collaborated to develop Chimera, an advanced artificial intelligence (AI) framework designed to enhance the accuracy and scalability of retrosynthesis prediction.
The Genesis of Chimera
The collaboration between Microsoft and Novartis was formalized in September 2019, with the announcement of a multiyear alliance aimed at transforming medicine through artificial intelligence. This partnership sought to leverage data and AI to revolutionize the discovery, development, and commercialization of medicines. A cornerstone of this initiative was the establishment of the Novartis AI Innovation Lab, which empowered scientists to utilize AI across various facets of their operations.
Building upon this foundation, researchers from Microsoft Research, Novartis Biomedical Research, and Jagiellonian University embarked on developing Chimera. This ensemble framework integrates multiple machine learning models, each with distinct inductive biases, to collaboratively predict retrosynthetic pathways. By combining the strengths of these diverse models, Chimera aims to overcome the limitations of traditional methods, particularly in accurately predicting rare and complex chemical reactions.
Key Features of Chimera
Ensemble Learning Approach: Chimera employs an ensemble of models, each trained with different inductive biases, to enhance prediction accuracy. This approach allows the framework to capture a broader range of chemical transformations, including those that are less common or previously underrepresented in training datasets.
Diverse Model Integration: The framework integrates various machine learning architectures, such as graph neural networks and sequence-to-sequence models, to process chemical data from multiple perspectives. This diversity enables Chimera to effectively handle the complexities inherent in chemical reactions and molecular structures.
Learned Ranking Mechanism: Chimera incorporates a learned ranking mechanism to synthesize outputs from its constituent models. This mechanism prioritizes the most plausible synthetic routes, thereby enhancing the reliability of the predictions and facilitating the selection of optimal pathways for chemical synthesis.
Scalability and Efficiency: Designed to handle large and complex datasets, Chimera offers scalability that is essential for modern chemical research. Its efficient processing capabilities enable researchers to explore a vast array of synthetic routes, accelerating the discovery and development of new compounds.
Impact on Chemical Synthesis
The introduction of Chimera marks a significant advancement in the field of chemical synthesis. By leveraging AI to predict retrosynthetic pathways with greater accuracy and scalability, Chimera addresses several critical challenges:
Enhanced Prediction Accuracy: The ensemble approach and diverse model integration enable Chimera to predict both common and rare chemical reactions more accurately, thereby expanding the scope of feasible synthetic routes.
Accelerated Drug Discovery: In pharmaceutical research, Chimera's capabilities can expedite the design and synthesis of new drugs by providing reliable retrosynthetic pathways, potentially reducing the time and cost associated with drug development.
Advancement in Materials Science: For materials science, Chimera's predictive power can facilitate the creation of novel materials with desired properties, opening new avenues for innovation in various industries.
Future Prospects
The development of Chimera represents a promising step toward the integration of AI in chemical synthesis. As the framework continues to evolve, it holds the potential to further transform the landscape of chemical research by:
Expanding Reaction Databases: By incorporating a broader range of chemical reactions into its training datasets, Chimera can enhance its predictive capabilities, particularly for novel and complex reactions.
Improving Model Interpretability: Advancements in AI interpretability could provide chemists with deeper insights into the reasoning behind Chimera's predictions, fostering greater trust and facilitating the validation of synthetic routes.
Integrating with Experimental Platforms: Future iterations of Chimera may integrate with experimental platforms, enabling real-time feedback and iterative refinement of synthetic pathways, thereby creating a more dynamic and responsive approach to chemical synthesis.
In summary, Chimera exemplifies the transformative potential of AI in chemical synthesis. Through its collaborative development by Microsoft and Novartis, it addresses longstanding challenges in retrosynthesis prediction, offering a powerful tool for researchers and paving the way for future innovations in the field.
For a visual overview of the collaboration between Novartis and Microsoft in leveraging AI for medical advancements, you may find the following video informative:
The Chimera Framework
In the field of chemical synthesis, accurately predicting retrosynthetic pathways is crucial for the efficient design and development of new molecules. Traditional methods often face challenges due to the complexity and diversity of chemical reactions. To address these challenges, Chimera, a collaborative AI framework developed by Microsoft and Novartis, employs an ensemble approach that integrates multiple models with diverse inductive biases. This strategy significantly enhances the accuracy and scalability of retrosynthesis prediction.
Understanding Ensemble Learning
Ensemble learning is a machine learning paradigm that combines the predictions of multiple models to achieve superior performance compared to individual models. The underlying principle is that diverse models, each capturing different aspects of the data, can collectively provide more accurate and robust predictions. This approach is particularly effective in complex domains like chemical synthesis, where single models may struggle to capture the full range of chemical reactions and molecular structures.
Chimera's Ensemble Framework
Chimera's ensemble framework integrates outputs from multiple machine learning models, each with distinct inductive biases. Inductive biases refer to the assumptions and prior knowledge that guide a model's learning process. By combining models with complementary biases, Chimera can leverage the strengths of each, leading to more accurate and reliable retrosynthesis predictions.
Diverse Inductive Biases in Chimera
The models integrated into Chimera are designed with diverse inductive biases to capture various facets of chemical reactions:
Edit Rule-Based Models: These models utilize predefined reaction templates and rules to predict retrosynthetic pathways. They are effective for common reactions but may have limitations with rare or novel reactions.
De-Novo Generation Models: Employing advanced architectures like Transformers, these models generate novel synthetic routes without relying on predefined templates. They are capable of predicting a broader range of reactions, including those that are less common or previously underrepresented.
Learning-Based Ensembling Strategy
Chimera employs a learning-based ensembling strategy to combine the predictions from its diverse models. This strategy involves training a meta-model to assign appropriate weights to the outputs of each base model, effectively synthesizing their predictions into a final, more accurate result. This approach allows Chimera to adaptively integrate the strengths of each model, improving overall prediction accuracy.
Advantages of Chimera's Ensemble Approach
The ensemble approach in Chimera offers several advantages:
Enhanced Accuracy: By integrating diverse models, Chimera can more accurately predict both common and rare chemical reactions, addressing the limitations of individual models.
Scalability: The framework's design allows it to handle large and complex datasets, making it suitable for modern chemical research that requires processing vast amounts of data.
Robustness: Combining models with different biases reduces the risk of overfitting to specific patterns in the data, leading to more generalizable and reliable predictions.
Impact on Chemical Synthesis
Chimera's ensemble approach has significant implications for chemical synthesis:
Accelerated Drug Discovery: By providing more accurate retrosynthetic pathways, Chimera can expedite the design and synthesis of new pharmaceutical compounds, potentially reducing the time and cost associated with drug development.
Advancement in Materials Science: The framework's capabilities can facilitate the creation of novel materials with desired properties, opening new avenues for innovation in various industries.
Future Prospects
The success of Chimera's ensemble approach suggests that future developments in chemical AI might benefit from similarly combining multiple specialized models rather than pursuing a one-size-fits-all solution. As the field progresses, integrating diverse models with complementary biases could lead to even more accurate and efficient predictive tools for chemical synthesis.
In summary, Chimera's ensemble approach, which integrates models with diverse inductive biases through a learning-based ensembling strategy, represents a significant advancement in retrosynthesis prediction. This methodology enhances the accuracy, scalability, and robustness of chemical synthesis planning, offering a powerful tool for researchers and paving the way for future innovations in the field.
Key Components of Chimera: NeuralLoc and R-SMILES 2
Chimera, the collaborative AI framework developed by Microsoft and Novartis, integrates two state-of-the-art models to enhance the accuracy and scalability of retrosynthesis prediction: NeuralLoc and R-SMILES 2. Each model contributes unique capabilities, addressing specific challenges in chemical synthesis.
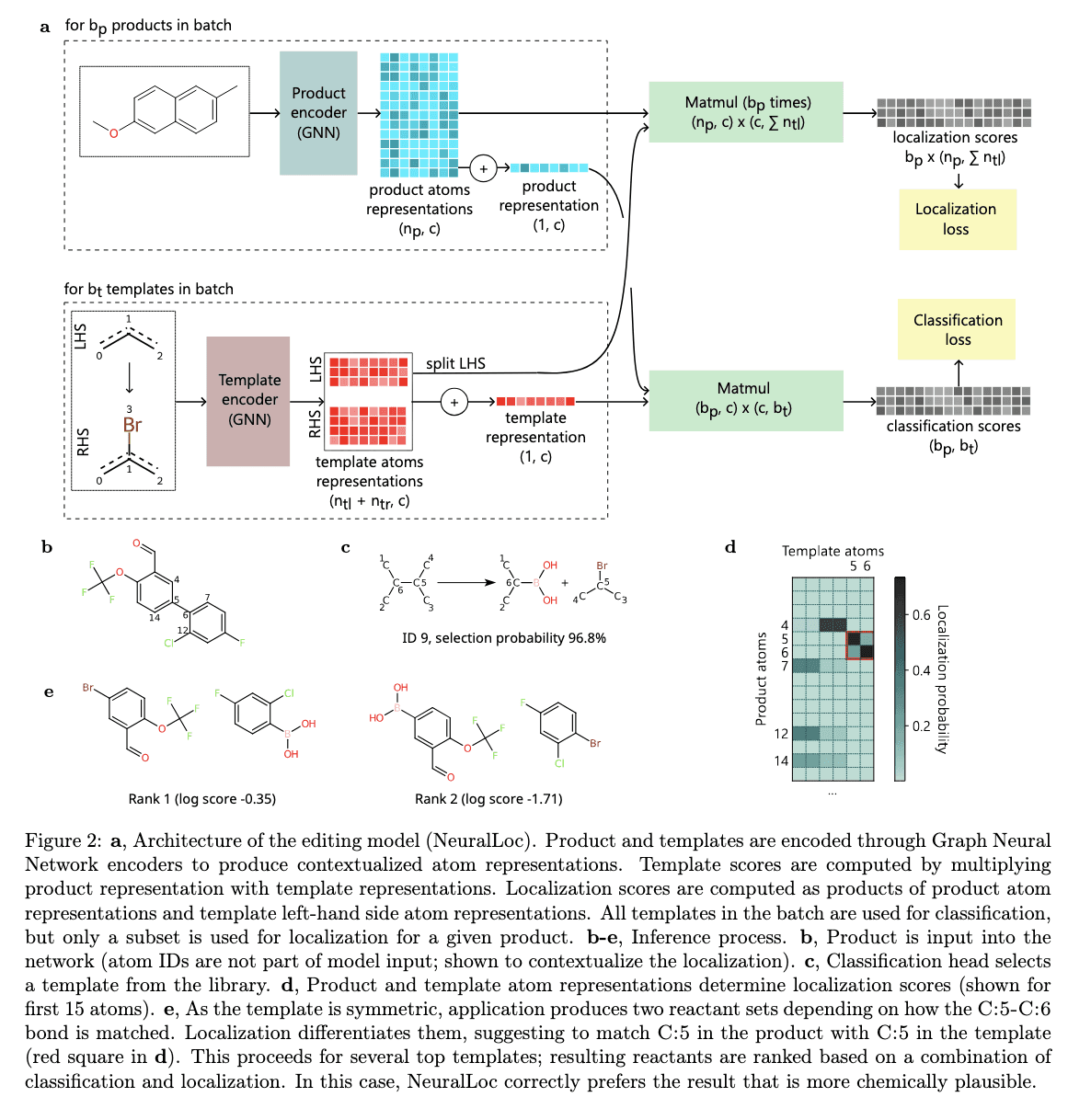
NeuralLoc: Molecule Editing with Graph Neural Networks
NeuralLoc is a pivotal component of Chimera, focusing on molecule editing through the application of graph neural networks (GNNs). GNNs are a class of neural networks designed to process data represented as graphs, making them particularly well-suited for modeling molecular structures, which are inherently graph-based.
Graph Neural Networks in Chemistry
In the context of chemistry, GNNs are employed to model molecules as graphs, where atoms are nodes and chemical bonds are edges. This representation allows GNNs to capture the complex relationships and interactions within molecular structures. By learning from large datasets of molecular structures and their properties, GNNs can predict various chemical properties and behaviors, including reactivity and stability.
NeuralLoc's Approach to Molecule Editing
NeuralLoc leverages GNNs to predict reaction sites and templates, facilitating the editing of molecular structures. This capability is crucial for retrosynthesis prediction, as it enables the identification of potential synthetic routes by determining where and how molecules can be modified. By encoding molecular structures as graphs, NeuralLoc can precisely predict reaction sites and templates, ensuring that predicted transformations align closely with known chemical rules while maintaining computational efficiency.
Advantages of NeuralLoc's Graph-Based Approach
The use of GNNs in NeuralLoc offers several advantages:
Precision in Reaction Site Prediction: By modeling molecules as graphs, NeuralLoc can accurately identify specific atoms and bonds involved in chemical reactions, leading to more precise retrosynthetic predictions.
Alignment with Chemical Rules: The graph-based representation allows NeuralLoc to incorporate chemical rules and principles directly into its learning process, ensuring that predicted reactions are chemically plausible.
Computational Efficiency: GNNs are capable of processing large and complex molecular datasets efficiently, making NeuralLoc suitable for high-throughput retrosynthesis prediction.
Impact on Retrosynthesis Prediction
By focusing on molecule editing through GNNs, NeuralLoc enhances Chimera's ability to predict both common and rare chemical reactions. This capability is particularly valuable in pharmaceutical and materials science, where the design of novel compounds often requires the synthesis of complex molecules with specific properties. NeuralLoc's precise prediction of reaction sites and templates enables researchers to explore a broader range of synthetic routes, accelerating the development of new compounds.
R-SMILES 2: De-Novo Generation with Advanced Sequence-to-Sequence Architectures
R-SMILES 2 is a pivotal component of Chimera, the collaborative AI framework developed by Microsoft and Novartis, designed to enhance the accuracy and scalability of retrosynthesis prediction. This model focuses on de-novo generation of reactant molecules using advanced sequence-to-sequence (seq2seq) architectures, specifically tailored to process SMILES (Simplified Molecular Input Line Entry System) strings.
Understanding De-Novo Generation in Chemical Synthesis
De-novo generation refers to the process of designing new molecules from scratch, without relying on existing templates or prior knowledge of their structures. In chemical synthesis, this approach is invaluable for discovering novel compounds with desired properties, such as potential pharmaceutical agents or materials with specific functionalities. The ability to predict retrosynthetic pathways through de-novo generation accelerates the drug discovery process and material innovation.
Sequence-to-Sequence Architectures in R-SMILES 2
R-SMILES 2 employs advanced sequence-to-sequence architectures to model the transformation of product molecules back to their reactant forms. These architectures are adept at handling the sequential nature of SMILES strings, which represent molecular structures as linear sequences of characters. The seq2seq model consists of two main components:
Encoder: The encoder processes the input SMILES string of the product molecule, converting it into a fixed-size latent representation that captures the essential features of the molecular structure.
Decoder: The decoder takes this latent representation and generates the corresponding SMILES string of the reactant molecule, effectively predicting the synthetic route in reverse.
This architecture enables R-SMILES 2 to learn complex mappings between product and reactant structures, facilitating accurate retrosynthesis predictions.
Architecture and Training of R-SMILES 2
The architecture of R-SMILES 2 is designed to handle the unique challenges of chemical structure representation and transformation. The input product is converted into a SMILES string and tokenized into a sequence of tokens. Before processing, sinusoidal positional embeddings are incorporated to infuse positional information, allowing the model to understand the spatial arrangement of atoms and bonds. The sequence then undergoes transformation through layers composed of grouped multi-query attention, RMS normalization, and feedforward layers with SwiGLU activations. The autoregressive decoder predicts the SMILES sequence of reactants utilizing self-attention over already produced tokens and cross-attention over encoder output. The model is trained using a cross-entropy loss function, optimizing it to accurately reconstruct reactant structures from product inputs.
Advantages of R-SMILES 2 in Retrosynthesis Prediction
R-SMILES 2 offers several advantages in the field of retrosynthesis prediction:
Template-Free Generation: Unlike traditional template-based methods, R-SMILES 2 does not rely on predefined reaction templates, allowing it to predict novel and diverse synthetic routes.
Handling Complex Structures: The seq2seq architecture is capable of managing the complexity of chemical structures, including those with intricate ring systems and stereochemistry, which are often challenging for other models.
Scalability: R-SMILES 2 can process large datasets efficiently, making it suitable for high-throughput screening in drug discovery and materials science.
Performance and Impact
In evaluations on the USPTO-50K dataset, R-SMILES 2 demonstrated significant improvements over baseline models, achieving a top-1 accuracy of 56.9% and a top-5 accuracy of 86.9%. These results underscore its effectiveness in accurately predicting retrosynthetic pathways.
The integration of R-SMILES 2 into Chimera enhances the framework's ability to predict both common and rare chemical reactions, thereby accelerating the design and synthesis of new compounds. This capability is particularly valuable in pharmaceutical and materials science, where the development of novel compounds with specific properties is essential.
Learning-to-Rank Strategy: Integrating Model Outputs with Precision
Chimera, the collaborative AI framework developed by Microsoft and Novartis, addresses the complexities of retrosynthesis prediction by employing a sophisticated learning-to-rank (LTR) strategy. This approach effectively integrates outputs from its constituent models—NeuralLoc and R-SMILES 2—ensuring that the most accurate and relevant synthetic pathways are prioritized.
Understanding Learning-to-Rank in Retrosynthesis Prediction
In the context of retrosynthesis prediction, a learning-to-rank strategy involves training a model to assign scores to potential synthetic routes based on their likelihood of success. By learning from historical data, the model can discern which features and patterns are indicative of successful reactions. This enables Chimera to rank multiple predicted pathways, highlighting those with the highest probability of yielding the desired product.
Mechanics of Chimera's Learning-to-Rank Strategy
Chimera's LTR strategy operates through the following key steps:
Model Output Generation: NeuralLoc and R-SMILES 2 independently generate a set of potential synthetic routes for a given target molecule.
Feature Extraction: For each predicted pathway, Chimera extracts a comprehensive set of features, including reaction conditions, reagent types, and molecular descriptors.
Ranking Model Training: Using a dataset of known reactions and their outcomes, Chimera trains a ranking model to assign scores to each pathway based on its features.
Pathway Ranking: The trained model ranks the predicted pathways, allowing Chimera to select the most promising routes for further exploration.
Advantages of the Learning-to-Rank Strategy
Implementing a learning-to-rank strategy offers several advantages:
Enhanced Accuracy: By learning from empirical data, Chimera can more accurately predict successful synthetic routes, reducing the reliance on predefined templates.
Adaptability: The LTR model can be continuously updated with new data, allowing Chimera to adapt to emerging trends and novel reaction conditions.
Prioritization of Resources: By ranking pathways, Chimera enables researchers to focus on the most promising synthetic routes, optimizing resource allocation and experimental efforts.
Impact on Retrosynthesis Prediction
The integration of a learning-to-rank strategy significantly enhances Chimera's ability to predict both common and rare chemical reactions. This capability is particularly valuable in pharmaceutical and materials science, where the design of novel compounds often requires the synthesis of complex molecules with specific properties. Chimera's precise ranking of synthetic routes accelerates the development of new compounds by highlighting the most viable pathways for experimental validation.
Performance and Validation
Chimera, the collaborative AI framework developed by Microsoft and Novartis, has undergone rigorous benchmark testing to assess its efficacy in retrosynthesis prediction. Utilizing established datasets such as USPTO-50K and USPTO-FULL, Chimera's performance has been evaluated against both common and rare chemical reactions, demonstrating its superior accuracy and robustness.
USPTO-50K Dataset: A Standard Benchmark
The USPTO-50K dataset comprises 50,000 chemical reactions extracted from U.S. patents, serving as a standard benchmark for evaluating retrosynthesis prediction models. These reactions are categorized into 10 distinct reaction classes, providing a diverse set of challenges for predictive models.
Chimera's Performance on USPTO-50K
In evaluations on the USPTO-50K dataset, Chimera demonstrated significant improvements over baseline models. The framework achieved a top-10 exact match accuracy of 56.9%, surpassing previous state-of-the-art methods. This performance underscores Chimera's capability to accurately predict synthetic routes for a wide range of chemical reactions.
USPTO-FULL Dataset: A Comprehensive Evaluation
The USPTO-FULL dataset offers a more extensive collection of chemical reactions, encompassing a broader spectrum of reaction types and complexities. This dataset serves as a comprehensive evaluation tool for assessing the generalization capabilities of retrosynthesis prediction models.
Chimera's Performance on USPTO-FULL
When tested on the USPTO-FULL dataset, Chimera continued to exhibit superior performance. The framework achieved a top-10 exact match accuracy of 55.6%, further validating its robustness across diverse reaction types.
Comparative Analysis with Baseline Models
Chimera's performance was compared to several baseline models, including NeuralLoc and R-SMILES 2. While each of these models demonstrated strong individual performance, Chimera's ensemble approach, integrating outputs from both NeuralLoc and R-SMILES 2, resulted in enhanced accuracy. This integration allowed Chimera to set new benchmarks for top-10 exact match accuracy on both USPTO-50K and USPTO-FULL datasets.
Implications for Chemical Synthesis
The superior performance of Chimera on these benchmark datasets signifies a substantial advancement in the field of retrosynthesis prediction. By accurately predicting both common and rare chemical reactions, Chimera facilitates the design of novel synthetic routes, thereby accelerating the development of new compounds in pharmaceutical and materials science.
Expert Evaluation: Organic Chemists' Insights on Chimera's Predictions
Chimera, the collaborative AI framework developed by Microsoft and Novartis, has undergone rigorous evaluations by organic chemists to assess its practical applicability in chemical synthesis. These expert assessments have highlighted Chimera's superior performance in predicting synthetic routes, particularly when compared to individual models.
Comparative Studies with Organic Chemists
In a series of evaluations, organic chemists were presented with synthetic routes predicted by Chimera alongside those generated by individual models. The results consistently favored Chimera's predictions, with experts preferring them over those from the standalone models. This preference underscores Chimera's effectiveness in providing accurate and reliable synthetic pathways that align with expert expectations.
Insights from Chemists
Chemists have noted that Chimera's ensemble approach, which integrates outputs from multiple models, offers a more comprehensive and nuanced prediction of synthetic routes. This integration captures a broader range of reaction possibilities, facilitating the identification of optimal pathways for complex chemical syntheses. The ability to predict both common and rare reactions with high accuracy has been particularly appreciated, as it enhances the efficiency of the synthesis planning process.
Implications for Chemical Synthesis
The positive evaluations from organic chemists indicate that Chimera is a valuable tool in the field of chemical synthesis. Its capacity to generate accurate and diverse synthetic routes accelerates the development of new compounds, benefiting areas such as pharmaceutical research and materials science. By aligning AI-driven predictions with expert insights, Chimera bridges the gap between computational models and practical laboratory applications.
Real-World Application: Chimera's Integration with Novartis' Internal Datasets
Chimera, the collaborative AI framework developed by Microsoft and Novartis, has demonstrated exceptional performance when applied to internal datasets at Novartis. This real-world application underscores Chimera's robustness and its capacity to generalize across diverse chemical reaction data.
Application to Novartis' Internal Datasets
To evaluate Chimera's practical utility, it was tested on an internal dataset comprising over 10,000 chemical reactions. This dataset, sourced from Novartis' extensive research archives, includes a wide array of reaction types and complexities, providing a comprehensive challenge for retrosynthesis prediction models. In this zero-shot setting—where no additional fine-tuning was performed—Chimera demonstrated superior accuracy compared to its constituent models. This outcome highlights Chimera's capability to generalize effectively across different datasets and predict viable synthetic pathways even in real-world scenarios.
Robustness Under Distribution Shifts
The evaluation also tested Chimera's robustness under distribution shifts, a common challenge in real-world applications where training and testing data distributions may differ. Chimera's performance remained consistent, further validating its reliability and adaptability in diverse settings.
Implications for Chemical Synthesis at Novartis
The successful application of Chimera to Novartis' internal datasets signifies a substantial advancement in computational chemistry. By accurately predicting synthetic routes for a wide range of chemical reactions, Chimera accelerates the development of new compounds, enhancing the efficiency of pharmaceutical research and materials science. Its ability to generalize across various datasets ensures that Chimera can be a valuable tool in diverse research contexts within Novartis.
Implications for Chemical Synthesis
The pharmaceutical industry is continually seeking innovative methods to expedite the development of new drugs, aiming to reduce the traditionally lengthy and costly process. Artificial intelligence (AI) has emerged as a transformative force in this endeavor, particularly in the realm of retrosynthetic route prediction. Chimera, a collaborative AI framework developed by Microsoft and Novartis, exemplifies this advancement by providing accurate retrosynthetic routes, thereby accelerating drug discovery.
The Traditional Drug Discovery Process
Traditionally, the drug discovery process involves several stages:
Target Identification and Validation: Determining and confirming biological molecules (typically proteins) that are involved in a disease process.
Hit Discovery: Identifying small molecules that interact with the target.
Lead Optimization: Refining these molecules to improve efficacy, selectivity, and pharmacokinetic properties.
Preclinical and Clinical Development: Testing the optimized compounds for safety and efficacy in laboratory and clinical settings.
This process can span over a decade and incur costs exceeding $1 billion.
The Impact of AI on Drug Discovery
AI has revolutionized various aspects of drug discovery, notably in:
Predicting Molecular Properties: AI models can forecast the biological activity, toxicity, and pharmacokinetics of compounds, enabling the identification of promising candidates early in the discovery phase.
Designing Novel Compounds: Generative models can create new molecular structures with desired properties, facilitating the development of drugs targeting previously undruggable proteins.
Optimizing Synthetic Routes: AI-driven retrosynthesis prediction models suggest efficient pathways for synthesizing target molecules, reducing the time and resources required for chemical synthesis.
Chimera's Contribution to Accelerating Drug Discovery
Chimera enhances the drug discovery process through:
Accurate Retrosynthetic Predictions: By leveraging an ensemble of models with diverse inductive biases, Chimera provides reliable synthetic routes for complex molecules, streamlining the synthesis of drug candidates.
Integration with Existing Databases: Chimera's ability to process and analyze large chemical datasets allows researchers to identify novel compounds and optimize existing ones more efficiently.
Facilitating De Novo Drug Design: Chimera's advanced sequence-to-sequence architectures enable the generation of new molecular structures with desired properties, expediting the design of potential therapeutics.
Case Studies Demonstrating Chimera's Efficacy
In practical applications, Chimera has demonstrated its capabilities:
Internal Datasets at Novartis: When applied to Novartis' proprietary datasets, Chimera accurately predicted synthetic routes for novel compounds, showcasing its robustness and generalization capabilities.
Benchmark Testing: Chimera's performance on datasets like USPTO-50K and USPTO-FULL has been exemplary, highlighting its superior accuracy in predicting both common and rare reactions.
Challenges and Future Directions
Despite its advancements, challenges remain:
Data Quality and Diversity: The effectiveness of AI models like Chimera depends on the quality and diversity of the training data. Ensuring comprehensive and representative datasets is crucial for accurate predictions.
Integration into Existing Workflows: Seamlessly incorporating AI tools into traditional drug discovery pipelines requires overcoming technical and organizational hurdles.
Regulatory Considerations: The use of AI in drug development must align with regulatory standards, necessitating ongoing collaboration between AI developers and regulatory bodies.
Looking ahead, the integration of AI frameworks like Chimera is poised to further streamline drug discovery, potentially reducing development timelines and costs, and enabling the creation of more effective and personalized therapeutics.
Advancements in artificial intelligence (AI) are revolutionizing material science, particularly in the design and synthesis of novel materials. AI frameworks like Chimera, developed collaboratively by Microsoft and Novartis, are at the forefront of this transformation, offering innovative solutions to traditional challenges in material synthesis planning.
Traditional Challenges in Material Synthesis
Historically, the development of new materials has been a labor-intensive process, relying heavily on empirical experimentation and serendipity. Researchers often faced difficulties in predicting the properties of new materials and devising efficient synthesis routes. This trial-and-error approach not only extended development timelines but also increased costs and resource consumption.
AI's Role in Transforming Material Synthesis
AI technologies are addressing these challenges by:
Predicting Material Properties: Machine learning models can analyze vast datasets to forecast the properties of new materials, enabling researchers to identify promising candidates more efficiently. This predictive capability accelerates the discovery phase and reduces the reliance on extensive experimental testing.
Optimizing Synthesis Routes: AI frameworks like Chimera utilize advanced algorithms to suggest optimal pathways for synthesizing target materials. By considering various reaction conditions and potential intermediates, these models propose efficient and cost-effective synthesis strategies. This optimization not only streamlines the synthesis process but also enhances the scalability of material production.
Facilitating Autonomous Synthesis: Integrating AI with automated laboratory systems allows for autonomous material synthesis. AI-driven platforms can design experiments, execute them, and analyze results with minimal human intervention, significantly accelerating the development cycle. For instance, autonomous synthesis of thin-film materials has been achieved by combining pulsed laser deposition with machine learning algorithms, enabling rapid exploration of material properties.
Chimera's Impact on Material Science
Chimera exemplifies the application of AI in material science by:
Enhancing Design Capabilities: By accurately predicting synthetic routes and material properties, Chimera empowers researchers to design materials with specific characteristics tailored to desired applications. This precision in design is particularly valuable in fields such as electronics, energy storage, and biomaterials.
Accelerating Discovery Processes: Chimera's ability to rapidly generate and evaluate potential materials expedites the discovery phase, reducing the time from concept to application. This acceleration is crucial in industries where time-to-market is a competitive advantage.
Supporting Sustainable Practices: By optimizing synthesis routes and predicting material properties, Chimera contributes to more sustainable material production processes. Efficient synthesis reduces waste and energy consumption, aligning with global sustainability goals.
Real-World Applications and Future Prospects
The integration of AI frameworks like Chimera into material science is already yielding tangible benefits:
Carbon Capture Materials: Amazon has piloted an AI-designed material for carbon removal in its data centers. Developed by Orbital Materials using AI, this CO₂-specific sponge operates with atomic-level precision, offering a cost-effective solution for carbon capture. This initiative underscores the potential of AI in developing materials for environmental sustainability.
Advanced Metamaterials: The Chimera metamaterial represents a significant breakthrough, capable of rendering objects invisible across microwave, visible light, and infrared wavelengths. This advancement has profound implications for optics and materials science, demonstrating the power of AI in creating novel materials with unique properties.
Looking ahead, the continued integration of AI in material science is expected to:
Enable Personalized Materials: AI will facilitate the design of materials tailored to specific applications, leading to more efficient and effective products across various industries.
Advance Sustainable Manufacturing: AI-driven optimization of synthesis routes will contribute to more sustainable manufacturing practices, reducing environmental impact and resource consumption.
Foster Interdisciplinary Collaboration: The convergence of AI, material science, and other disciplines will drive innovation, leading to the development of materials with unprecedented properties and applications.
In conclusion, AI frameworks like Chimera are revolutionizing material science by enhancing synthesis planning, accelerating discovery processes, and supporting sustainable practices. As AI technologies continue to evolve, their integration into material science promises to unlock new possibilities, leading to the creation of innovative materials that address complex challenges across various sectors.
Conclusion
Chimera, developed collaboratively by Microsoft and Novartis, represents a significant advancement in the field of retrosynthesis prediction. By integrating diverse models with complementary inductive biases, Chimera has achieved notable milestones that have enhanced the accuracy and scalability of predicting synthetic routes for complex molecules.
1. Superior Accuracy in Retrosynthesis Prediction
Chimera has demonstrated exceptional performance in predicting retrosynthetic pathways, particularly for reactions that are less frequent yet crucial in chemical synthesis. Traditional machine learning models often struggled with these rare reactions, leading to lower prediction accuracy. Chimera's ensemble approach, combining multiple models with diverse inductive biases, has addressed this challenge effectively. In benchmark tests, Chimera achieved a top-1 accuracy of 71.5% on the USPTO-50K dataset, surpassing the performance of individual models.
2. Excellence in Multi-Step Retrosynthesis Tasks
Beyond single-step reactions, Chimera has excelled in multi-step retrosynthesis tasks, achieving close to 100% success rates on benchmarks such as SimpRetro. This capability is particularly valuable for complex molecules that require multi-step synthetic routes, highlighting Chimera's potential to transform computational retrosynthesis.
3. Robust Generalization Across Diverse Datasets
Chimera's robustness is evident in its ability to generalize across various datasets. When tested on an internal dataset from a major pharmaceutical company, Chimera demonstrated superior accuracy compared to its constituent models, even without additional fine-tuning. This adaptability underscores Chimera's capability to handle distribution shifts and perform effectively in real-world scenarios.
4. Preference Among Expert Chemists
In evaluations conducted with PhD-level organic chemists, Chimera's predictions were preferred over those from baseline models in terms of quality. This preference indicates that Chimera's predictions align more closely with expert judgment, enhancing its practical utility in chemical synthesis planning.
5. Integration of Diverse Modeling Approaches
Chimera's architecture integrates two primary modeling approaches:
Editing Model: Utilizes a transformer-based architecture to modify molecular graphs directly, identifying and adjusting reaction centers while preserving the overall molecular structure.
De-Novo Model: Employs a sequence-to-sequence approach, treating molecules as SMILES strings, to generate reactant predictions from scratch, allowing for more flexible and creative solutions to synthesis problems.
This combination of editing and de-novo synthesis strategies enables Chimera to handle a wide range of chemical reactions and synthetic routes.
6. Scalability and Efficiency
Chimera's ensemble strategy, which uses a weighted voting mechanism to combine predictions from both models, is designed for scalability. The weights are dynamically adjusted based on confidence scores and historical performance patterns, allowing Chimera to efficiently process large datasets and complex chemical spaces.
7. Contribution to Sustainable Chemistry
By providing accurate and efficient retrosynthetic routes, Chimera contributes to more sustainable practices in chemical manufacturing. Optimized synthesis routes reduce waste and energy consumption, aligning with global sustainability goals and promoting greener chemistry practices.
8. Acceleration of Drug Discovery and Material Science
Chimera's capabilities have significant implications for drug discovery and material science. By accurately predicting synthetic routes, Chimera accelerates the development of new pharmaceuticals and facilitates the creation of novel materials with desired properties. This acceleration is crucial in responding to emerging health challenges and in the development of personalized medicine.
Press contact
Timon Harz
oneboardhq@outlook.com
Other posts





Company
About
Blog
Careers
Press
Legal
Privacy
Terms
Security